- Record: found
- Abstract: found
- Article: found
PREP-Mt: predictive RNA editor for plant mitochondrial genes
Read this article at
Abstract
Background
In plants, RNA editing is a process that converts specific cytidines to uridines and uridines to cytidines in transcripts from virtually all mitochondrial protein-coding genes. There are thousands of plant mitochondrial genes in the sequence databases, but sites of RNA editing have not been determined for most. Accurate methods of RNA editing site prediction will be important in filling in this information gap and could reduce or even eliminate the need for experimental determination of editing sites for many sequences. Because RNA editing tends to increase protein conservation across species by "correcting" codons that specify unconserved amino acids, this principle can be used to predict editing sites by identifying positions where an RNA editing event would increase the conservation of a protein to homologues from other plants. PREP-Mt takes this approach to predict editing sites for any protein-coding gene in plant mitochondria.
Results
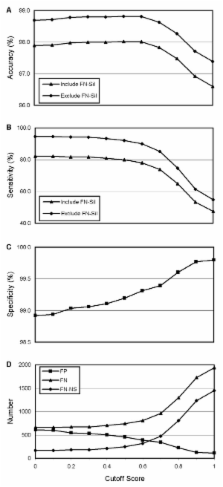
To test the general applicability of the PREP-Mt methodology, RNA editing sites were predicted for 370 full-length or nearly full-length DNA sequences and then compared to the known sites of RNA editing for these sequences. Of 60,263 cytidines in this test set, PREP-Mt correctly classified 58,994 as either an edited or unedited site (accuracy = 97.9%). PREP-Mt properly identified 3,038 of the 3,698 known sites of RNA editing (sensitivity = 82.2%) and 55,956 of the 56,565 known unedited sites (specificity = 98.9%). Accuracy and sensitivity increased to 98.7% and 94.7%, respectively, after excluding the 489 silent editing sites (which have no effect on protein sequence or function) from the test set.
Conclusion
These results indicate that PREP-Mt is effective at identifying C to U RNA editing sites in plant mitochondrial protein-coding genes. Thus, PREP-Mt should be useful in predicting protein sequences for use in molecular, biochemical, and phylogenetic analyses. In addition, PREP-Mt could be used to determine functionality of a mitochondrial gene or to identify particular sequences with unusual editing properties. The PREP-Mt methodology should be applicable to any system where RNA editing increases protein conservation across species.
Related collections
Most cited references38
- Record: found
- Abstract: found
- Article: not found
Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs.
- Record: found
- Abstract: found
- Article: not found
Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA.
- Record: found
- Abstract: found
- Article: not found