- Record: found
- Abstract: found
- Article: found
Clinical and GAA gene mutation analysis in mainland Chinese patients with late-onset Pompe disease: identifying c.2238G > C as the most common mutation
Read this article at
Abstract
Background
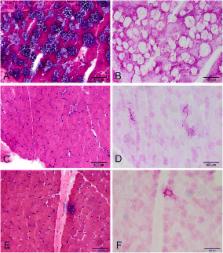
Pompe disease is an autosomal recessive lysosomal glycogen storage disorder that has been reported in different ethnic populations which carry different common mutations of the acid alpha-glucosidase ( GAA) gene. The GAA mutation pattern in mainland Chinese patients with late-onset Pompe disease is still not well understood.
Methods
We presented the clinical and genetic characteristics of 27 mainland Chinese late-onset Pompe patients from 24 families.
Results
GAA mutation analysis revealed 26 different mutations, including 10 that were novel. The allelic frequency of c.2238G > C (p.W746C) was found to be 27.08% in this patient group. Respiratory dysfunction was diagnosed in 10 of 11 patients who underwent pulmonary function evaluation, although only four required ventilator support at night.
Conclusions
Our findings indicate that c.2238G > C (p.W746C) is the most common mutation in mainland Chinese late-onset Pompe patients, as observed in Taiwanese patients. The novel mutations identified in this study expand the genetic spectrum of late-onset Pompe disease, and the prevalence of respiratory dysfunction highlights the importance of monitoring pulmonary function in late-onset Pompe patients.
Related collections
Most cited references28
- Record: found
- Abstract: found
- Article: not found
Late onset Pompe disease: clinical and neurophysiological spectrum of 38 patients including long-term follow-up in 18 patients.
- Record: found
- Abstract: found
- Article: not found