- Record: found
- Abstract: found
- Article: found
Genetic interaction network has a very limited impact on the evolutionary trajectories in continuous culture-grown populations of yeast
Read this article at
Abstract
Background
The impact of genetic interaction networks on evolution is a fundamental issue. Previous studies have demonstrated that the topology of the network is determined by the properties of the cellular machinery. Functionally related genes frequently interact with one another, and they establish modules, e.g., modules of protein complexes and biochemical pathways. In this study, we experimentally tested the hypothesis that compensatory evolutionary modifications, such as mutations and transcriptional changes, occur frequently in genes from perturbed modules of interacting genes.
Results
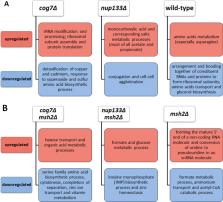
Using Saccharomyces cerevisiae haploid deletion mutants as a model, we investigated two modules lacking COG7 or NUP133, which are evolutionarily conserved genes with many interactions. We performed laboratory evolution experiments with these strains in two genetic backgrounds (with or without additional deletion of MSH2), subjecting them to continuous culture in a non-limiting minimal medium. Next, the evolved yeast populations were characterized through whole-genome sequencing and transcriptome analyses. No obvious compensatory changes resulting from inactivation of genes already included in modules were identified. The supposedly compensatory inactivation of genes in the evolved strains was only rarely observed to be in accordance with the established fitness effect of the genetic interaction network. In fact, a substantial majority of the gene inactivations were predicted to be neutral in the experimental conditions used to determine the interaction network. Similarly, transcriptome changes during continuous culture mostly signified adaptation to growth conditions rather than compensation of the absence of the COG7, NUP133 or MSH2 genes . However, we noticed that for genes whose inactivation was deleterious an upregulation of transcription was more common than downregulation.
Conclusions
Our findings demonstrate that the genetic interactions and the modular structure of the network described by others have very limited effects on the evolutionary trajectory following gene deletion of module elements in our experimental conditions and has no significant impact on short-term compensatory evolution. However, we observed likely compensatory evolution in functionally related (albeit non-interacting) genes.
Related collections
Most cited references86

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools

- Record: found
- Abstract: found
- Article: found
fastp: an ultra-fast all-in-one FASTQ preprocessor
- Record: found
- Abstract: found
- Article: not found