- Record: found
- Abstract: found
- Article: found
Molecular Docking and Molecular Dynamics (MD) Simulation of Human Anti-Complement Factor H (CFH) Antibody Ab42 and CFH Polypeptide
Read this article at
Abstract
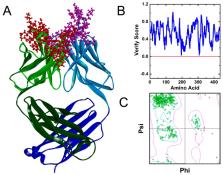
An understanding of the interaction between the antibody and its targeted antigen and knowing of the epitopes are critical for the development of monoclonal antibody drugs. Complement factor H (CFH) is implied to play a role in tumor growth and metastasis. An autoantibody to CHF is associated with anti-tumor cell activity. The interaction of a human monoclonal antibody Ab42 that was isolated from a cancer patient with CFH polypeptide (pCFH) antigen was analyzed by molecular docking, molecular dynamics (MD) simulation, free energy calculation, and computational alanine scanning (CAS). Experimental alanine scanning (EAS) was then carried out to verify the results of the theoretical calculation. Our results demonstrated that the Ab42 antibody interacts with pCFH by hydrogen bonds through the Tyr315, Ser100, Gly33, and Tyr53 residues on the complementarity-determining regions (CDRs), respectively, with the amino acid residues of Pro441, Ile442, Asp443, Asn444, Ile447, and Thr448 on the pCFH antigen. In conclusion, this study has explored the mechanism of interaction between Ab42 antibody and its targeted antigen by both theoretical and experimental analysis. Our results have important theoretical significance for the design and development of relevant antibody drugs.
Related collections
Most cited references34
- Record: found
- Abstract: found
- Article: not found
Complement control protein factor H: the good, the bad, and the inadequate.
- Record: found
- Abstract: found
- Article: not found
ZRANK: reranking protein docking predictions with an optimized energy function.
- Record: found
- Abstract: found
- Article: not found