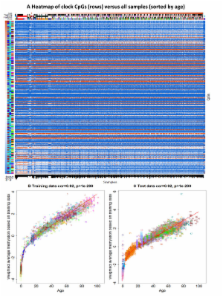
Introduction DNA methylation is an epigenetic mechanism that plays an important role in gene expression regulation, development, and disease. Increasing evidence points to the distinct contributions of genetic [1], [2], [3], [4], [5], environmental [6], [7], [8], and stochastic factors to DNA methylation levels at individual genomic regions. In addition, DNA methylation patterns at specific CpG-sites can also vary over time within an individual [9], [10] and correspondingly, age-related methylation changes have been identified in multiple tissues and organisms [11], [12], [13], [14], [15]. Although age-related changes in methylation have been implicated in healthy ageing and longevity, the causes and functional consequences of these remain unclear. Ageing is a complex process, which represents the progression of multiple degenerative processes within an individual. Studies in different organisms have identified many factors that contribute to lifespan and the rate of healthy ageing within an individual. These include components of biological mechanisms involved in cellular senescence, oxidative stress, DNA repair, protein glycation, and others (see [16]). Taking these into account, the concept of biological age has been proposed as a better predictor of lifespan and functional capacity than chronological age alone. Previous studies have proposed that certain traits can be used as measures of biological age [17] and have put forward a stringent definition of an ageing biomarker (see [18]). Here, we examined age-related phenotypes that have previously been considered biomarkers of ageing (see [19]), specifically white cell telomere length, blood pressure, lung function, grip strength, bone mineral density, parental longevity, parental age at reproduction, and serum levels of 5-dehydroepiandrosterone (DHEAS), cholesterol, albumin, and creatinine. Epigenetic studies of age-related phenotypes can help identify molecular changes that associate with the ageing process. Such changes may include both biological markers of accumulated stochastic damage in the organism, as well as specific susceptibility factors that may play a regulatory role. We explored the hypothesis that epigenetic changes contribute to the rate of ageing and potential longevity in a sample of 172 middle-aged female twins, where methylation profiles and age-DMRs were previously characterized in 93 individuals from the sample [14]. We compared DNA methylation patterns with chronological age in the sample of 172 individuals and related epigenetic variation to age-related phenotypes that have previously been used as biomarkers of ageing. We identified phenotype-associated DNA methylation changes and combined genetic, epigenetic, expression, and phenotype data to help understand the underlying mechanism of association between epigenetic variation, chronological age, and ageing-related traits. Results DNA methylation patterns in twins associate with genetic variants We characterized DNA methylation patterns in a sample of 172 female twins at 26,690 promoter CpG-sites that map uniquely across the genome. We observed that the majority of autosomal CpG-sites were un-methylated (beta 5% and an Impute info value of >0.8. Altogether, there were 2,054,344 directly genotyped and imputed autosomal SNPs used in the QTL analyses. Gene expression data Gene expression estimates and eQTLs from lymphoblastoid cell lines (LCLs) in the samples were obtained for 168 individuals in the study [21]. Gene expression levels were measured using the Illumina expression array HumanHT-12 version 3 as previously described [21]. Each sample had three technical replicates and log2 - transformed expression signals were quantile normalized first across the 3 replicates of each individual, followed by quantile normalization across all individuals [21]. We assigned methylation and expression probes to the gene with the nearest transcription start site using Refseq gene annotations. For each gene we obtained the mean methylation (or gene expression) estimate, by averaging values over multiple methylation (or gene expression) probes if more than one probe was assigned to that gene. There were altogether 435 genes nearest to the 490 age DMRs, of which 348 had transcription start sites within 2 kb of the methylation CpG-sites and for which we also had whole blood methylation data and LCLs gene expression data in 168 individuals. Linear mixed effects models and Spearman rank correlations were used to compare methylation and expression data per gene. Methylation QTL analyses We tested for methylation QTLs at 24,522 autosomal probes, which had at least one SNP within 50 kb of the probe that passed genotype QC criteria. We fitted a linear mixed-effects model, regressing the methylation levels at each probe on fixed-effect terms including genotype, methylation chip, and sample order on the methylation chip, and random-effect terms denoting family structure and zygosity. Prior to these analyses, the methylation values at each CpG-site were normalized to N(0,1). Results from meQTL analyses are presented at a false discovery rate (FDR) of 5%, estimated by permutation. Here, we permuted the methylation data at the 24,522 autosomal probes, performed cis association analyses on the permuted and normalized methylation data, and repeated this procedure for 10 replicates selecting the most associated SNP per probe per replicate. FDR was calculated as the fraction of significant hits in the permuted data compared to the observed data at each p-value threshold. DMR analyses Linear mixed effects models were used to assess evidence for DMRs. In the a-DMR analyses we regressed the raw methylation levels at each probe on fixed-effect terms including age, methylation chip, and sample order on the methylation chip, and random-effect terms denoting family structure and zygosity. To assess the significance of the a-DMRs we compared this model to a null model, which excluded age from the fixed-effects terms. In the ap-DMR analyses we regressed the raw methylation levels at each probe on fixed-effect terms including phenotype, methylation chip, and sample order on chip, and random-effect for family and zygosity, and compared the fit of this model to a null model which excluded the phenotype. We also performed the ap-DMR analyses by including age as a fixed effect covariate in both the null and alternative models. We also repeated both the a-DMR and ap-DMR analyses using normalized methylation levels (to N(0,1)) and observed that the reported DMRs were top-ranked in the normalized analyses. To assess genome-wide significance we performed 100 permutations and estimated FDR by calculating the fraction of significant hits in the permuted data compared to the observed data at a specific P-value threshold. Monozygotic twin DMR effects were calculated in the set of 21 MZ twin pairs where both twins were assayed within the same batch of methylation arrays. We estimated MZ-DMRs for 12 phenotypes where data were available in at least 12 MZ pairs. For each phenotype of interest we fitted a linear model comparing phenotype within-pair differences to methylation within-pair differences and reported the P-values obtained from the F-statistics from the overall regression. For the age-corrected analyses we fitted the regression including age as a covariate and compared the results to a null model, which included phenotype differences and age alone. We performed 100 replicates to estimate FDR 5% significant results as described above. At the FDR 5% significance threshold (nominal P = 2.03×10−6), we estimated 35% power to detect the observed correlation (Pearson correlation = 0.83) between methylation MZ-differences at cg01136458 in CSMD1 (mean MZ-beta-difference = 5%) and LDL MZ-differences (mean MZ-LDL-difference = 0.73 SD) in 20 MZ pairs. Age DMR replication The replication sample comprised 44 MZ twins discordant for psychosis, that were profiled on the Illumina 27K array as previously described [27]. The sample consisted of younger adults (age range 20–61, median age 28), including both female and male twin pairs. We compared methylation against age at the 490 a-DMRs both in the entire set of 44 twins and in the set of 22 unaffected unrelated individuals. In the set of 44 twins we fitted linear mixed effect models, regressing the normalized beta values per probe (normalized to N(0,1)) against methylation chip, sample order on the chip, sex, and age as fixed effects, and family as random effect. In the set of 22 unaffected unrelated individuals comprising the control twin from each pair we calculated Spearman rank correlation coefficients on the untransformed methylation beta values against age. Genome-wide association scans Genome-wide association scans were performed using linear mixed effects models for 12 phenotypes including telomere length, systolic blood pressure (SBP), diastolic blood pressure (DBP), FEV1 and FVC to examine lung function, grip strength, bone mineral density (BMD), serum levels of DHEAS, serum total cholesterol levels, serum high density cholesterol levels (HDL), calculated levels of serum low density cholesterol (LDL), serum albumin levels, and serum creatinine levels. Linear models were fit as described in the meQTL analyses section substituting phenotype for methylation, using an additive model. SNPs with evidence for association that surpassed P = 0.001, were considered in the overlap across cis-meQTL, genotype-phenotype, and DMR findings. Functional characterization of DMRs The 26,690 methylation probes were assigned to CpG islands according to previous definitions [54], resulting in 11,299 CpG sites that were in CpG islands and 15,391 that were outside of CpG islands. Histone modification ChIP-seq data were obtained from the Encode project from one CEPH HapMap LCL (GM12878) in the UCSC genome browser. Peaks in the genome-wide read-depth distribution from ChIP-seq histone modifications H3K9ac, H3K27ac, H3K27me3, H3K4me1, H3K4me2, and H3K4me3 were obtained as previously described (see [1]). Enrichment a-DMR estimates were calculated as the proportion of a-DMRs in each functional category (CpG islands or histone peaks) over the proportion of 26,690 probe in that functional category. Enrichment 95% confidence intervals were estimated using bootstrap percentile intervals of 1,000 re-samplings of the a-DMR data per annotation category. Gene ontology term enrichment analysis was performed using the GOrilla tool for identifying enriched GO terms in the ranked list of a-DMR genes [31], using Refseq gene annotations in the entire set of 26,690 probes as background. Supporting Information Figure S1 Summary characteristics of DNA methylation patterns in 172 female twins. Distribution of methylation scores (beta) in (A) autosomal and (B) X-chromosomal probes in all individuals. (PDF) Click here for additional data file. Figure S2 Distribution of intra-class correlation coefficients (ICC) in twins. Density plots of ICC in MZ twins (red) and DZ twins (blue) for two batches of methylation data (batch 1 consists of 93 twins (left) and batch 2 consists of 79 twins (right)). The mean MZ-ICCs and DZ-ICCs were estimated as 0.257 and 0.168 in batch 1 (MZ-ICC vs DZ-ICC P<2×10−16), and as 0.3557 and 0.261 in batch 2 (MZ-ICC vs DZ-ICC P<2×10−16). The corresponding methylation probe heritabilities were calculated as 2(ICC_MZ - ICC_DZ) and the genome-wide estimates were 0.176 (95%CI:0.168–0.185) and 0.188 (95%CI:0.180–0.196) for the data in batch 1 (left) and batch 2 (right), respectively. (PDF) Click here for additional data file. Figure S3 Correlation across age-related phenotypes. Below diagonal plots represent each pair of phenotypes and the corresponding rank correlation coefficient is shown above the diagonal. (PDF) Click here for additional data file. Figure S4 EWAS results for age-related phenotypes. FDR 5% ap-DMRs were obtained for (A) LDL, (B) lung function (FVC), and (C) maternal longevity (MLONG) with (green) and without (blue) age-correction. Red dashed lines correspond to age-corrected (A) and non-age-corrected (B,C) analysis FDR 5% levels. (PDF) Click here for additional data file. Figure S5 Lack of enrichment of age-related phenotype DMR association in the set of age DMRs. (PDF) Click here for additional data file. Figure S6 Evidence for co-methylation. Spearman correlation in methylation levels between all pair-wise CpG-sites (black) and between a-DMR CpG-sites (red) in the sample of 172 related individuals (solid line) and a subset of 96 unrelated individuals (dotted line). (PDF) Click here for additional data file. Table S1 List of 490 a-DMRs. (XLS) Click here for additional data file. Table S2 Descriptive statistics of the age-related phenotypes. (XLS) Click here for additional data file. Table S3 List of 19 a-DMRs associated with proportion of lymphocytes. (XLS) Click here for additional data file. Table S4 Overlap across genotype-methylation (cis-meQTLs), methylation-phenotype (ap-DMRs), and genotype-phenotype (GWAS) association results. (XLS) Click here for additional data file. Table S5 JASPAR motif search results in the set of a-DMR genes. Results are shown at P = 0.05 threshold. (XLS) Click here for additional data file. Table S6 Gene Ontology term enrichment results in the set of a-DMR genes. GO term enrichment in a-DMR genes was assessed relative to the background set of 14,344 genes that map nearest to the 26,690 probes tested. Results are shown at P = 1e-6 for biological processes and molecular functions. (XLS) Click here for additional data file.