
- Record: found
- Abstract: found
- Article: found
Modeling radiation injury-induced cell death and countermeasure drug responses in a human Gut-on-a-Chip
Read this article at
Abstract
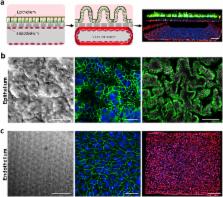
Studies on human intestinal injury induced by acute exposure to γ-radiation commonly rely on use of animal models because culture systems do not faithfully mimic human intestinal physiology. Here we used a human Gut-on-a-Chip (Gut Chip) microfluidic device lined by human intestinal epithelial cells and vascular endothelial cells to model radiation injury and assess the efficacy of radiation countermeasure drugs in vitro. Exposure of the Gut Chip to γ-radiation resulted in increased generation of reactive oxygen species, cytotoxicity, apoptosis, and DNA fragmentation, as well as villus blunting, disruption of tight junctions, and compromise of intestinal barrier integrity. In contrast, pre-treatment with a potential prophylactic radiation countermeasure drug, dimethyloxaloylglycine (DMOG), significantly suppressed all of these injury responses. Thus, the human Gut Chip may serve as an in vitro platform for studying radiation-induced cell death and associate gastrointestinal acute syndrome, in addition to screening of novel radio-protective medical countermeasure drugs.
Related collections
Most cited references57
- Record: found
- Abstract: found
- Article: not found
Intestinal Goblet Cells and Mucins in Health and Disease: Recent Insights and Progress
- Record: found
- Abstract: found
- Article: not found
Tumor response to radiotherapy regulated by endothelial cell apoptosis.
- Record: found
- Abstract: found
- Article: not found