- Record: found
- Abstract: found
- Article: found
The molecular landscape of neural differentiation in the developing Drosophila brain revealed by targeted scRNA-seq and multi-informatic analysis

Read this article at
SUMMARY
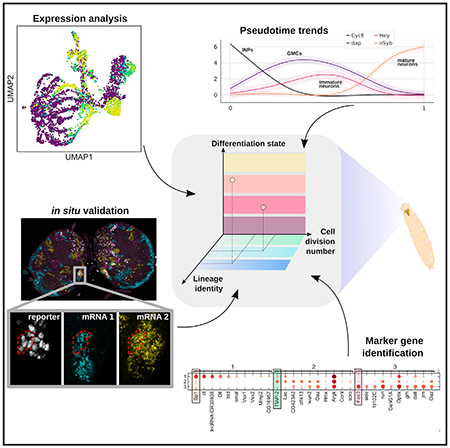
The Drosophila type II neuroblast lineages present an attractive model to investigate the neurogenesis and differentiation process as they adapt to a process similar to that in the human outer subventricular zone. We perform targeted single-cell mRNA sequencing in third instar larval brains to study this process of the type II NB lineage. Combining prior knowledge, in silico analyses, and in situ validation, our multi-informatic investigation describes the molecular landscape from a single developmental snapshot. 17 markers are identified to differentiate distinct maturation stages. 30 markers are identified to specify the stem cell origin and/or cell division numbers of INPs, and at least 12 neuronal subtypes are identified. To foster future discoveries, we provide annotated tables of pairwise gene-gene correlation in single cells and MiCV, a web tool for interactively analyzing scRNA-seq datasets. Taken together, these resources advance our understanding of the neural differentiation process at the molecular level.
In brief
Using a combination of targeted scRNA-seq, in situ RNA staining, and a multi-informatic analysis paradigm, Michki et al. characterize the transcriptome landscape of thousands of type II neurons and their progenitors in the developing larval fruit fly brain.
Graphical Abstract
Related collections
Most cited references81
- Record: found
- Abstract: found
- Article: not found
Fiji: an open-source platform for biological-image analysis.
- Record: found
- Abstract: found
- Article: not found
STAR: ultrafast universal RNA-seq aligner.
- Record: found
- Abstract: found
- Article: not found