- Record: found
- Abstract: found
- Article: found
Variant in the X-chromosome spliceosomal gene GPKOW causes male-lethal microcephaly with intrauterine growth restriction
Read this article at
Abstract
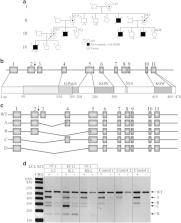
Congenital microcephaly, with or without additional developmental defects, is a heterogeneous disorder resulting from impaired brain development during early fetal life. The majority of causative genetic variants identified thus far are inherited in an autosomal recessive manner and impact key cellular pathways such as mitosis, DNA damage response and repair, apoptosis and splicing. Here, we report a novel donor splice site variant in the G-patch domain and KOW motifs ( GPKOW) gene (NG_021310.2:g.6126G>A, NM_015698.4:c.331+5G>A) that segregates with affected and carrier status in a multigenerational family with an X-linked perinatal lethal condition characterized by severe microcephaly and intrauterine growth restriction (IUGR). GPKOW is a core member of the spliceosome that has been shown in numerous model organisms and in human cells to be essential for survival. By investigating GPKOW transcripts in lymphoblastoid cell lines (LCLs) of three carrier females, we show that the GPKOW c.331+5G>A variant disrupts normal splicing of its pre-mRNAs. In a clonal culture expressing only the c.331+5G>A allele isolated from one carrier female LCL, we observed an 80% reduction in wild type GPKOW mRNA, 70% reduction in the full length GPKOW protein and the presence of a truncated GPKOW protein with possible dominant negative effect. Based on our and published data we propose that the GPKOW gene is essential for fetal development and when disrupted, leads to a severe, male-lethal phenotype characterised by microcephaly and IUGR.
Related collections
Most cited references20
- Record: found
- Abstract: found
- Article: not found
Analysis of protein-coding genetic variation in 60,706 humans
- Record: found
- Abstract: found
- Article: not found
A general framework for estimating the relative pathogenicity of human genetic variants
- Record: found
- Abstract: found
- Article: not found