- Record: found
- Abstract: found
- Article: found
All SNPs Are Not Created Equal: Genome-Wide Association Studies Reveal a Consistent Pattern of Enrichment among Functionally Annotated SNPs
Read this article at
Abstract
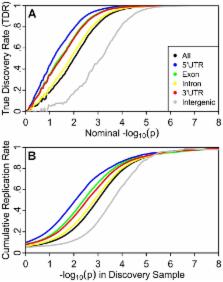
Recent results indicate that genome-wide association studies (GWAS) have the potential to explain much of the heritability of common complex phenotypes, but methods are lacking to reliably identify the remaining associated single nucleotide polymorphisms (SNPs). We applied stratified False Discovery Rate (sFDR) methods to leverage genic enrichment in GWAS summary statistics data to uncover new loci likely to replicate in independent samples. Specifically, we use linkage disequilibrium-weighted annotations for each SNP in combination with nominal p-values to estimate the True Discovery Rate (TDR = 1−FDR) for strata determined by different genic categories. We show a consistent pattern of enrichment of polygenic effects in specific annotation categories across diverse phenotypes, with the greatest enrichment for SNPs tagging regulatory and coding genic elements, little enrichment in introns, and negative enrichment for intergenic SNPs. Stratified enrichment directly leads to increased TDR for a given p-value, mirrored by increased replication rates in independent samples. We show this in independent Crohn's disease GWAS, where we find a hundredfold variation in replication rate across genic categories. Applying a well-established sFDR methodology we demonstrate the utility of stratification for improving power of GWAS in complex phenotypes, with increased rejection rates from 20% in height to 300% in schizophrenia with traditional FDR and sFDR both fixed at 0.05. Our analyses demonstrate an inherent stratification among GWAS SNPs with important conceptual implications that can be leveraged by statistical methods to improve the discovery of loci.
Author Summary
Modern genome-wide association studies (GWAS) have failed to identify large portions of the genetic basis of common, complex traits. Recent work suggested this could be because many genetic variants, each with individually small effects, compose their genetic architecture, limiting the power of GWAS. Moreover, these variants appear more abundantly in and near genes. Using genome annotations, summary statistics from several of the largest GWAS, and established statistical methods for quantifying distributions of test statistics, we show a consistency across studies. Namely, we show that, across all assessed traits, the test statistics resulting from SNPs that are related to the 5′ UTR of genes show the largest abundance of associations, while SNPs related to exons and the 3′UTR are also enriched. SNPs related to introns are only moderately enriched, and intergenic SNPs show a depletion of associations relative to the average SNP. This enrichment corresponds directly to increased replication across independent samples and can be incorporated a priori into statistical methods to improve discovery and prediction. Our results contribute to on-going debates about the functional nature of the genetic architecture of complex traits and point to avenues for leveraging existing GWAS data for discovery in future GWA and sequencing studies.
Related collections
Most cited references18
- Record: found
- Abstract: not found
- Article: not found
Evolution at two levels in humans and chimpanzees.
- Record: found
- Abstract: found
- Article: not found