- Record: found
- Abstract: found
- Article: found
Fixed-length haplotypes can improve genomic prediction accuracy in an admixed dairy cattle population
Read this article at
Abstract
Background
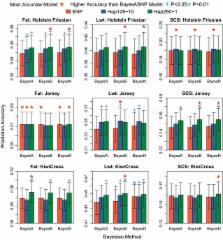
Fitting covariates representing the number of haplotype alleles rather than single nucleotide polymorphism (SNP) alleles may increase genomic prediction accuracy if linkage disequilibrium between quantitative trait loci and SNPs is inadequate. The objectives of this study were to evaluate the accuracy, bias and computation time of Bayesian genomic prediction methods that fit fixed-length haplotypes or SNPs. Genotypes at 37,740 SNPs that were common to Illumina BovineSNP50 and high-density panels were phased for ~58,000 New Zealand dairy cattle. Females born before 1 June 2008 were used for training, and genomic predictions for milk fat yield (n = 24,823), liveweight (n = 13,283) and somatic cell score (n = 24,864) were validated within breed (predominantly Holstein–Friesian, predominantly Jersey, or admixed KiwiCross) in later-born females. Covariates for haplotype alleles of five lengths (125, 250, 500 kb, 1 or 2 Mb) were generated and rare haplotypes were removed at four thresholds (1, 2, 5 or 10%), resulting in 20 scenarios tested. Genomic predictions fitting covariates for either SNPs or haplotypes were calculated by using BayesA, BayesB or BayesN. This is the first study to quantify the accuracy of genomic prediction using haplotypes across the whole genome in an admixed population.
Results
A correlation of 0.349 ± 0.016 between yield deviation and genomic breeding values was obtained for milk fat yield in Holstein–Friesians using BayesA fitting covariates. Genomic predictions were more accurate with short haplotypes than with SNPs but less accurate with longer haplotypes than with SNPs. Fitting only the most frequent haplotype alleles reduced computation time with little decrease in prediction accuracy for short haplotypes. Trends were similar for all traits and breeds and there was little difference between Bayesian methods.
Conclusions
Fitting covariates for haplotype alleles rather than SNPs can increase prediction accuracy, although it decreased drastically for long (>500 kb) haplotypes. In this population, fitting 250 kb haplotypes with a 1% frequency threshold resulted in the highest genomic prediction accuracy and fitting 125 kb haplotypes with a 10% frequency threshold improved genomic prediction accuracy with comparable computation time to fitting SNPs. This increased accuracy is likely to increase genetic gain by changing the ranking of selection candidates.
Related collections
Most cited references39
- Record: found
- Abstract: not found
- Book: not found
Introduction to Quantitative Genetics
- Record: found
- Abstract: found
- Article: not found
Development and Characterization of a High Density SNP Genotyping Assay for Cattle
- Record: found
- Abstract: found
- Article: not found