- Record: found
- Abstract: found
- Article: not found
Global Analysis of Genetic, Epigenetic and Transcriptional Polymorphisms in Arabidopsis thaliana Using Whole Genome Tiling Arrays
Read this article at

Abstract
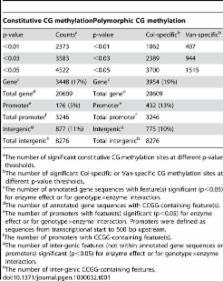
Whole genome tiling arrays provide a high resolution platform for profiling of genetic, epigenetic, and gene expression polymorphisms. In this study we surveyed natural genomic variation in cytosine methylation among Arabidopsis thaliana wild accessions Columbia (Col) and Vancouver (Van) by comparing hybridization intensity difference between genomic DNA digested with either methylation-sensitive ( HpaII) or -insensitive ( MspI) restriction enzyme. Single Feature Polymorphisms (SFPs) were assayed on a full set of 1,683,620 unique features of Arabidopsis Tiling Array 1.0F (Affymetrix), while constitutive and polymorphic CG methylation were assayed on a subset of 54,519 features, which contain a 5′CCGG3′ restriction site. 138,552 SFPs (1% FDR) were identified across enzyme treatments, which preferentially accumulated in pericentromeric regions. Our study also demonstrates that at least 8% of all analyzed CCGG sites were constitutively methylated across the two strains, while about 10% of all analyzed CCGG sites were differentially methylated between the two strains. Within euchromatin arms, both constitutive and polymorphic CG methylation accumulated in central regions of genes but under-represented toward the 5′ and 3′ ends of the coding sequences. Nevertheless, polymorphic methylation occurred much more frequently in gene ends than constitutive methylation. Inheritance of methylation polymorphisms in reciprocal F1 hybrids was predominantly additive, with F1 plants generally showing levels of methylation intermediate between the parents. By comparing gene expression profiles, using matched tissue samples, we found that magnitude of methylation polymorphism immediately upstream or downstream of the gene was inversely correlated with the degree of expression variation for that gene. In contrast, methylation polymorphism within genic region showed weak positive correlation with expression variation. Our results demonstrated extensive genetic and epigenetic polymorphisms between Arabidopsis accessions and suggested a possible relationship between natural CG methylation variation and gene expression variation.
Author Summary
The functional expression of DNA sequence depends on the chromatin status. Epigenetic marks at specific loci could affect local chromatin accessibility, thus affect the gene activity of that loci. We applied an enzyme methylome approach to globally detect one type of epigenetic mark, cytosine methylation at CCGG restriction sites. Simultaneous transcriptional profiling allowed gene expression differences to be compared with DNA methylation differences, suggesting functional regulatory regions. Our method reveals natural variation in chromatin patterns which may underlie phenotypic variation.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening.
- Record: found
- Abstract: found
- Article: not found
DNA methylation profiling of human chromosomes 6, 20 and 22
- Record: found
- Abstract: found
- Article: not found