- Record: found
- Abstract: found
- Article: found
Whole genome sequencing identified genomic diversity and candidated genes associated with economic traits in Northeasern Merino in China
Read this article at
Abstract
Introduction: Northeast Merino (NMS) is a breed developed in Northeast China during the 1960s for wool and meat production. It exhibits excellent traits such as high wool yield, superior meat quality, rapid growth rate, robust disease resistance, and adaptability to cold climates. However, no studies have used whole-genome sequencing data to investigate the superior traits of NMS.
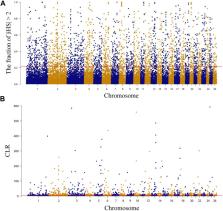
Methods: In this study, we investigated the population structure, genetic diversity, and selection signals of NMS using whole-genome sequencing data from 20 individuals. Two methods (integrated haplotype score and composite likelihood ratio) were used for selection signal analysis, and the Fixation Index was used to explore the selection signals of NMS and the other two breeds, Mongolian sheep and South African meat Merino.
Results: The results showed that NMS had low inbreeding levels, high genomic diversity, and a pedigree of both Merino breeds and Chinese local breeds. A total length of 14.09 Mb genomic region containing 287 genes was detected using the two methods. Further exploration of the functions of these genes revealed that they are mainly concentrated in wool production performance ( IRF2BP2, MAP3K7, and WNT3), meat production performance ( NDUFA9, SETBP1, ZBTB38, and FTO), cold resistance ( DNAJC13, LPGAT1, and PRDM16), and immune response ( PRDM2, GALNT8, and HCAR2). The selection signals of NMS and the other two breeds annotated 87 and 23 genes, respectively. These genes were also mainly focused on wool and meat production performance.
Conclusion: These results provide a basis for further breeding improvement, comprehensive use of this breed, and a reference for research on other breeds.
Related collections
Most cited references82

- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform
- Record: found
- Abstract: found
- Article: not found
MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms.
- Record: found
- Abstract: found
- Article: not found