- Record: found
- Abstract: found
- Article: found
The evolutionary dynamics of variant antigen genes in Babesia reveal a history of genomic innovation underlying host–parasite interaction
Read this article at
Abstract
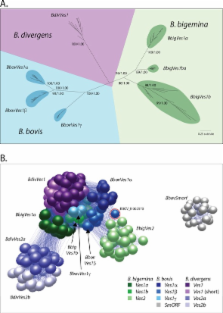
Babesia spp. are tick-borne, intraerythrocytic hemoparasites that use antigenic variation to resist host immunity, through sequential modification of the parasite-derived variant erythrocyte surface antigen (VESA) expressed on the infected red blood cell surface. We identified the genomic processes driving antigenic diversity in genes encoding VESA ( ves1) through comparative analysis within and between three Babesia species, ( B. bigemina, B. divergens and B. bovis). Ves1 structure diverges rapidly after speciation, notably through the evolution of shortened forms ( ves2) from 5′ ends of canonical ves1 genes. Phylogenetic analyses show that ves1 genes are transposed between loci routinely, whereas ves2 genes are not. Similarly, analysis of sequence mosaicism shows that recombination drives variation in ves1 sequences, but less so for ves2, indicating the adoption of different mechanisms for variation of the two families. Proteomic analysis of the B. bigemina PR isolate shows that two dominant VESA1 proteins are expressed in the population, whereas numerous VESA2 proteins are co-expressed, consistent with differential transcriptional regulation of each family. Hence, VESA2 proteins are abundant and previously unrecognized elements of Babesia biology, with evolutionary dynamics consistently different to those of VESA1, suggesting that their functions are distinct.
Related collections
Most cited references81
- Record: found
- Abstract: found
- Article: not found
MRBAYES: Bayesian inference of phylogenetic trees.

- Record: found
- Abstract: found
- Article: found
Toward almost closed genomes with GapFiller
- Record: found
- Abstract: found
- Article: not found