- Record: found
- Abstract: found
- Article: found
Single-Nuclei RNA Sequencing Assessment of the Hepatic Effects of 2,3,7,8-Tetrachlorodibenzo- p-dioxin
Read this article at
Abstract
Background and Aims
Characterization of cell specific transcriptional responses to hepatotoxicants is lost in the averages of bulk RNA-sequencing (RNA-seq). Single-cell/nuclei RNA-seq technologies enable the transcriptomes of individual cell (sub)types to be assessed within the context of in vivo models.
Methods
Single-nuclei RNA-sequencing (snSeq) of frozen liver samples from male C57BL/6 mice gavaged with sesame oil vehicle or 30 μg/kg 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) every 4 days for 28 days was used to demonstrate the application of snSeq for the evaluation of xenobiotics.
Results
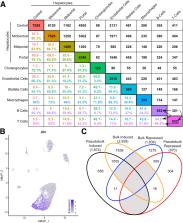
A total of 19,907 genes were detected across 16,015 nuclei from control and TCDD-treated livers. Eleven cell (sub)types reflected the expected cell diversity of the liver including distinct pericentral, midzonal, and periportal hepatocyte subpopulations. TCDD altered relative proportions of cell types and elicited cell-specific gene expression profiles. For example, macrophages increased from 0.5% to 24.7%, while neutrophils were only present in treated samples, consistent with histological evaluation. The number of differentially expressed genes (DEGs) in each cell type ranged from 122 (cholangiocytes) to 7625 (midcentral hepatocytes), and loosely correlated with the basal expression level of Ahr, the canonical mediator of TCDD and related compounds. In addition to the expected functions within each cell (sub)types, RAS signaling and related pathways were specifically enriched in nonparenchymal cells while metabolic process enrichment occurred primarily in hepatocytes. snSeq also identified the expansion of a Kupffer cell subtype highly expressing Gpnmb, as reported in a dietary NASH model.
Graphical abstract

Related collections
Most cited references51

- Record: found
- Abstract: found
- Article: found
GSVA: gene set variation analysis for microarray and RNA-Seq data
- Record: found
- Abstract: found
- Article: not found
Integrating single-cell transcriptomic data across different conditions, technologies, and species

- Record: found
- Abstract: found
- Article: found