- Record: found
- Abstract: found
- Article: found
SLAMF1 is required for TLR4-mediated TRAM-TRIF–dependent signaling in human macrophages

Read this article at
Abstract
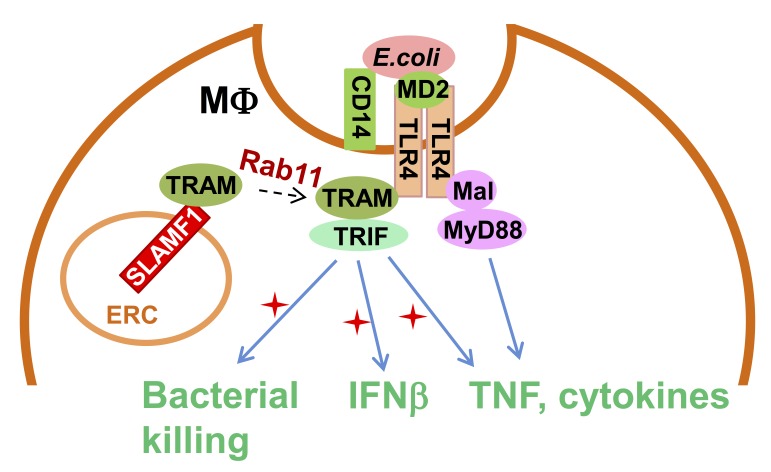
Yurchenko et al. discover that the Ig-like receptor molecule SLAMF1 enhances production of type I interferon induced by Gram-negative bacteria through modulation of MyD88-independent TLR4 signaling. This makes SLAMF1 a potential target for controlling inflammatory responses against Gram-negative bacteria.
Abstract
Signaling lymphocytic activation molecule family 1 (SLAMF1) is an Ig-like receptor and a costimulatory molecule that initiates signal transduction networks in a variety of immune cells. In this study, we report that SLAMF1 is required for Toll-like receptor 4 (TLR4)-mediated induction of interferon β (IFNβ) and for killing of Gram-negative bacteria by human macrophages. We found that SLAMF1 controls trafficking of the Toll receptor–associated molecule (TRAM) from the endocytic recycling compartment (ERC) to Escherichia coli phagosomes. In resting macrophages, SLAMF1 is localized to ERC, but upon addition of E. coli, it is trafficked together with TRAM from ERC to E. coli phagosomes in a Rab11-dependent manner. We found that endogenous SLAMF1 protein interacted with TRAM and defined key interaction domains as amino acids 68 to 95 of TRAM as well as 15 C-terminal amino acids of SLAMF1. Interestingly, the SLAMF1–TRAM interaction was observed for human but not mouse proteins. Overall, our observations suggest that SLAMF1 is a new target for modulation of TLR4–TRAM–TRIF inflammatory signaling in human cells.
Graphical Abstract
Related collections
Most cited references42
- Record: found
- Abstract: found
- Article: not found
TLR signaling augments macrophage bactericidal activity through mitochondrial ROS
- Record: found
- Abstract: found
- Article: not found
TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation.
- Record: found
- Abstract: found
- Article: not found