- Record: found
- Abstract: found
- Article: not found
Genome-Wide Association Study reveals genetic risk underlying Parkinson’s disease

Abstract
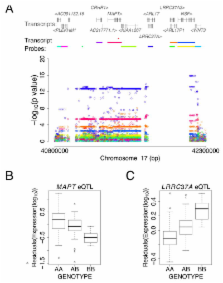
We performed a genome-wide association study (GWAS) in 1,713 Caucasian patients with Parkinson’s disease (PD) and 3,978 controls. After replication in 3,361 cases and 4,573 controls, two strong association signals were observed: in the α-synuclein gene( SNCA) (rs2736990, OR=1.23, p=2.24×10 −16) and at the MAPT locus (rs393152, OR=0.77, p=1.95×10 −16). We exchanged data with colleagues performing a GWAS in Asian PD cases. Association at SNCA was replicated in the Asian GWAS 1, confirming this as a major risk locus across populations. We were able to replicate the effect of a novel locus detected in the Asian cohort ( PARK16, rs823128, OR=0.66, p=7.29×10 −8) and provide evidence supporting the role of common variability around LRRK2 in modulating risk for PD (rs1491923, OR=1.14, p=1.55×10 −5). These data demonstrate an unequivocal role for common genetic variability in the etiology of typical PD and suggest population specific genetic heterogeneity in this disease.
Related collections
Most cited references11
- Record: found
- Abstract: found
- Article: not found
Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases.
- Record: found
- Abstract: found
- Article: not found
Genomewide association study for susceptibility genes contributing to familial Parkinson disease.
- Record: found
- Abstract: found
- Article: not found