- Record: found
- Abstract: found
- Article: found
Identification of epigenetic modifications that contribute to pathogenesis in therapy-related AML: Effective integration of genome-wide histone modification with transcriptional profiles
Read this article at
Abstract
Background
Therapy-related, secondary acute myeloid leukemia (t-AML) is an increasingly frequent complication of intensive chemotherapy. This malignancy is often characterized by abnormalities of chromosome 7, including large deletions or chromosomal loss. A variety of studies suggest that decreased expression of the EZH2 gene located at 7q36.1 is critical in disease pathogenesis. This histone methyltransferase has been implicated in transcriptional repression through modifying histone H3 on lysine 27 (H3k27). However, the critical target genes of EZH2 and their regulatory roles remain unclear.
Method
To characterize the subset of EZH2 target genes that might contribute to t-AML pathogenesis, we developed a novel computational analysis to integrate tissue-specific histone modifications and genome-wide transcriptional regulation. Initial integrative analysis utilized a novel "seq2gene" strategy to explore largely the target genes of chromatin immuneprecipitation sequencing (ChIP-seq) enriched regions. By combining seq2gene with our Phenotype-Genotype-Network (PGNet) algorithm, we enriched genes with similar expression profiles and genomic or functional characteristics into "biomodules".
Results
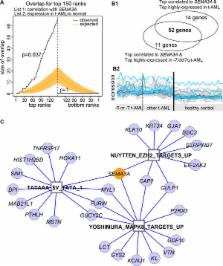
Initial studies identified SEMA3A (semaphoring 3A) as a novel oncogenic candidate that is regulated by EZH2-silencing, using data derived from both normal and leukemic cell lines as well as murine cells deficient in EZH2. A microsatellite marker at the SEMA3A promoter has been associated with chemosensitivity and radiosensitivity. Notably, our subsequent studies in primary t-AML demonstrate an expected up-regulation of SEMA3A that is EZH2-modulated. Furthermore, we have identified three biomodules that are co-expressed with SEMA3A and up-regulated in t-AML, one of which consists of previously characterized EZH2-repressed gene targets. The other two biomodules include MAPK8 and TATA box targets. Together, our studies suggest an important role for EZH2 targets in t-AML pathogenesis that warrants further study.
Related collections
Most cited references37
- Record: found
- Abstract: found
- Article: not found
Genomic Views of Distant-Acting Enhancers
- Record: found
- Abstract: found
- Article: not found