- Record: found
- Abstract: found
- Article: found
Dynamics of Rad9 Chromatin Binding and Checkpoint Function Are Mediated by Its Dimerization and Are Cell Cycle–Regulated by CDK1 Activity
Read this article at
Abstract
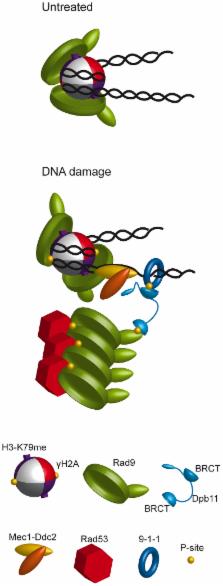
Saccharomyces cerevisiae Rad9 is required for an effective DNA damage response throughout the cell cycle. Assembly of Rad9 on chromatin after DNA damage is promoted by histone modifications that create docking sites for Rad9 recruitment, allowing checkpoint activation. Rad53 phosphorylation is also dependent upon BRCT-directed Rad9 oligomerization; however, the crosstalk between these molecular determinants and their functional significance are poorly understood. Here we report that, in the G1 and M phases of the cell cycle, both constitutive and DNA damage-dependent Rad9 chromatin association require its BRCT domains. In G1 cells, GST or FKBP dimerization motifs can substitute to the BRCT domains for Rad9 chromatin binding and checkpoint function. Conversely, forced Rad9 dimerization in M phase fails to promote its recruitment onto DNA, although it supports Rad9 checkpoint function. In fact, a parallel pathway, independent on histone modifications and governed by CDK1 activity, allows checkpoint activation in the absence of Rad9 chromatin binding. CDK1-dependent phosphorylation of Rad9 on Ser11 leads to specific interaction with Dpb11, allowing Rad53 activation and bypassing the requirement for the histone branch.
Author Summary
In response to DNA damage all eukaryotic cells activate a surveillance mechanism, known as the DNA damage checkpoint, which delays cell cycle progression and modulates DNA repair. Yeast RAD9 was the first DNA damage checkpoint gene identified. The genetic tools available in this model system allow to address relevant questions to understand the molecular mechanisms underlying the Rad9 biological function. By chromatin-binding and domain-swapping experiments, we found that Rad9 is recruited into DNA both in unperturbed and in DNA–damaging conditions, and we identified the molecular determinants required for such interaction. Moreover, the extent of chromatin-bound Rad9 is regulated during the cell cycle and influences its role in checkpoint activation. In fact, the checkpoint function of Rad9 in G1 cells is solely mediated by its interaction with modified histones, while in M phase it occurs through an additional scaffold protein, named Dpb11. Productive Rad9-Dpb11 interaction in M phase requires Rad9 phosphorylation by CDK1, and we identified the Ser11 residue as the major CDK1 target. The model of Rad9 action that we are presenting can be extended to other eukaryotic organisms, since Rad9 and Dpb11 have been conserved through evolution from yeast to mammalian cells.
Related collections
Most cited references73
- Record: found
- Abstract: found
- Article: not found
Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae.
- Record: found
- Abstract: found
- Article: not found
Regulation of DNA repair throughout the cell cycle.
- Record: found
- Abstract: found
- Article: not found