- Record: found
- Abstract: found
- Article: found
DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach
Read this article at
Abstract
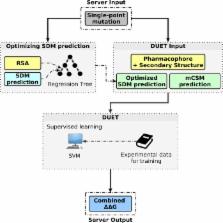
Cancer genome and other sequencing initiatives are generating extensive data on non-synonymous single nucleotide polymorphisms (nsSNPs) in human and other genomes. In order to understand the impacts of nsSNPs on the structure and function of the proteome, as well as to guide protein engineering, accurate in silicomethodologies are required to study and predict their effects on protein stability. Despite the diversity of available computational methods in the literature, none has proven accurate and dependable on its own under all scenarios where mutation analysis is required. Here we present DUET, a web server for an integrated computational approach to study missense mutations in proteins. DUET consolidates two complementary approaches (mCSM and SDM) in a consensus prediction, obtained by combining the results of the separate methods in an optimized predictor using Support Vector Machines (SVM). We demonstrate that the proposed method improves overall accuracy of the predictions in comparison with either method individually and performs as well as or better than similar methods. The DUET web server is freely and openly available at http://structure.bioc.cam.ac.uk/duet.
Related collections
Most cited references19
- Record: found
- Abstract: not found
- Article: not found
The interpretation of protein structures: estimation of static accessibility.
- Record: found
- Abstract: found
- Article: not found
Prediction of protein stability changes for single-site mutations using support vector machines.
- Record: found
- Abstract: found
- Article: not found