- Record: found
- Abstract: found
- Article: found
Comprehensive transcriptional variability analysis reveals gene networks regulating seed oil content of Brassica napus
Read this article at
Abstract
Background
Regulation of gene expression plays an essential role in controlling the phenotypes of plants. Brassica napus ( B. napus) is an important source for the vegetable oil in the world, and the seed oil content is an important trait of B. napus.
Results
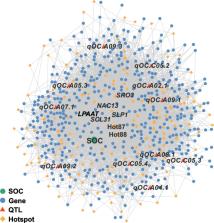
We perform a comprehensive analysis of the transcriptional variability in the seeds of B. napus at two developmental stages, 20 and 40 days after flowering (DAF). We detect 53,759 and 53,550 independent expression quantitative trait loci (eQTLs) for 79,605 and 76,713 expressed genes at 20 and 40 DAF, respectively. Among them, the local eQTLs are mapped to the adjacent genes more frequently. The adjacent gene pairs are regulated by local eQTLs with the same open chromatin state and show a stronger mode of expression piggybacking. Inter-subgenomic analysis indicates that there is a feedback regulation for the homoeologous gene pairs to maintain partial expression dosage. We also identify 141 eQTL hotspots and find that hotspot87-88 co-localizes with a QTL for the seed oil content. To further resolve the regulatory network of this eQTL hotspot, we construct the XGBoost model using 856 RNA-seq datasets and the Basenji model using 59 ATAC-seq datasets. Using these two models, we predict the mechanisms affecting the seed oil content regulated by hotspot87-88 and experimentally validate that the transcription factors, NAC13 and SCL31, positively regulate the seed oil content.
Related collections
Most cited references100

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools

- Record: found
- Abstract: found
- Article: found
BEDTools: a flexible suite of utilities for comparing genomic features
- Record: found
- Abstract: found
- Article: not found