- Record: found
- Abstract: found
- Article: found
Severe Phenotype in a Patient With Homozygous 15q21.2 Microdeletion Involving BCL2L10, GNB5, and MYO5C Genes, Resembling Infantile Developmental Disorder With Cardiac Arrhythmias (IDDCA)
Read this article at
Abstract
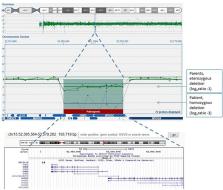
Homozygous and compound heterozygous mutations in GNB5 gene have been associated with a wide spectrum of clinical presentations, ranging from neurodevelopmental issues with or without cardiac arrhythmia (LADCI) to severe developmental delay with epileptic encephalopathy, retinal dystrophy, and heart rhythm abnormalities (IDDCA). While missense or missense/non-sense mutations usually lead to milder form, the biallelic loss of function of GNB5 gene causes the severe multisystemic IDDCA phenotype. So far, only 27 patients have been described with GNB5-associated disease. We report the first case of a patient carrying a homozygous 15q21.2 microdeletion, encompassing GNB5 and the two contiguous genes BCL2L10 and MYO5C. The clinical features of the child are consistent with the severe IDDCA phenotype, thus confirming the GNB5 loss-of-function mechanism in determining such presentation of the disease.
Related collections
Most cited references20
- Record: found
- Abstract: found
- Article: not found
Walking to work: roles for class V myosins as cargo transporters.
- Record: found
- Abstract: found
- Article: not found
Array CGH in patients with learning disability (mental retardation) and congenital anomalies: updated systematic review and meta-analysis of 19 studies and 13,926 subjects.
- Record: found
- Abstract: found
- Article: not found