- Record: found
- Abstract: found
- Article: found
Bi-allelic genetic variants in the translational GTPases GTPBP1 and GTPBP2 cause a distinct identical neurodevelopmental syndrome

Read this article at
Summary
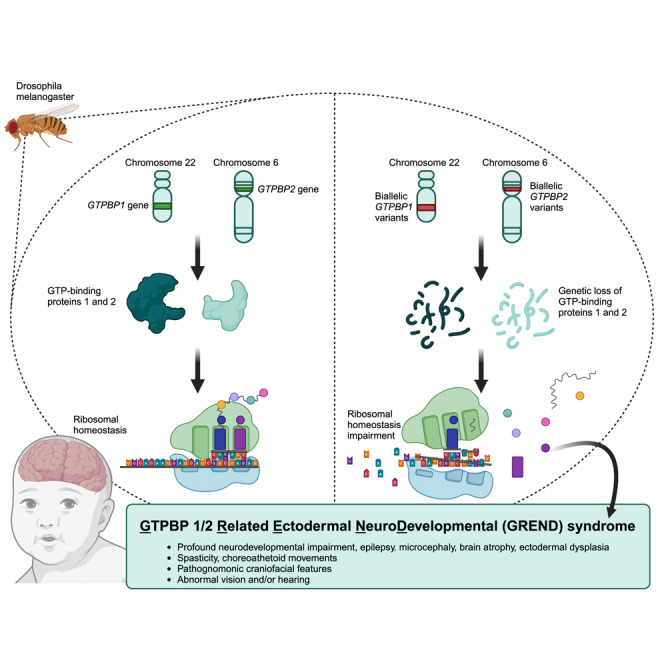
The homologous genes GTPBP1 and GTPBP2 encode GTP-binding proteins 1 and 2, which are involved in ribosomal homeostasis. Pathogenic variants in GTPBP2 were recently shown to be an ultra-rare cause of neurodegenerative or neurodevelopmental disorders (NDDs). Until now, no human phenotype has been linked to GTPBP1. Here, we describe individuals carrying bi-allelic GTPBP1 variants that display an identical phenotype with GTPBP2 and characterize the overall spectrum of GTP-binding protein (1/2)-related disorders. In this study, 20 individuals from 16 families with distinct NDDs and syndromic facial features were investigated by whole-exome (WES) or whole-genome (WGS) sequencing. To assess the functional impact of the identified genetic variants, semi-quantitative PCR, western blot, and ribosome profiling assays were performed in fibroblasts from affected individuals. We also investigated the effect of reducing expression of CG2017, an ortholog of human GTPBP1/2, in the fruit fly Drosophila melanogaster. Individuals with bi-allelic GTPBP1 or GTPBP2 variants presented with microcephaly, profound neurodevelopmental impairment, pathognomonic craniofacial features, and ectodermal defects. Abnormal vision and/or hearing, progressive spasticity, choreoathetoid movements, refractory epilepsy, and brain atrophy were part of the core phenotype of this syndrome. Cell line studies identified a loss-of-function (LoF) impact of the disease-associated variants but no significant abnormalities on ribosome profiling. Reduced expression of CG2017 isoforms was associated with locomotor impairment in Drosophila. In conclusion, bi-allelic GTPBP1 and GTPBP2 LoF variants cause an identical, distinct neurodevelopmental syndrome. Mutant CG2017 knockout flies display motor impairment, highlighting the conserved role for GTP-binding proteins in CNS development across species.
Graphical abstract
Abstract
Bi-allelic variants in the translational GTPases GTPBP1 and GTPBP2 may affect ribosomal translational control and impair brain development and neurological function. This research identifies 20 individuals with homozygous GTPBP1 and GTPBP2 variants leading to an identical neurodevelopmental syndrome, which we defined as “Gtpbp1/2-related ectodermal neurodevelopmental (GREND) syndrome.”
Related collections
Most cited references31

- Record: found
- Abstract: found
- Article: found
GENCODE reference annotation for the human and mouse genomes
- Record: found
- Abstract: found
- Article: not found