- Record: found
- Abstract: found
- Article: found
Identifying Mendelian disease genes with the Variant Effect Scoring Tool
Read this article at
Abstract
Background
Whole exome sequencing studies identify hundreds to thousands of rare protein coding variants of ambiguous significance for human health. Computational tools are needed to accelerate the identification of specific variants and genes that contribute to human disease.
Results
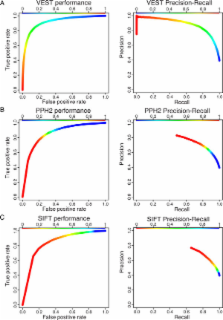
We have developed the Variant Effect Scoring Tool (VEST), a supervised machine learning-based classifier, to prioritize rare missense variants with likely involvement in human disease. The VEST classifier training set comprised ~ 45,000 disease mutations from the latest Human Gene Mutation Database release and another ~45,000 high frequency (allele frequency >1%) putatively neutral missense variants from the Exome Sequencing Project. VEST outperforms some of the most popular methods for prioritizing missense variants in carefully designed holdout benchmarking experiments (VEST ROC AUC = 0.91, PolyPhen2 ROC AUC = 0.86, SIFT4.0 ROC AUC = 0.84). VEST estimates variant score p-values against a null distribution of VEST scores for neutral variants not included in the VEST training set. These p-values can be aggregated at the gene level across multiple disease exomes to rank genes for probable disease involvement. We tested the ability of an aggregate VEST gene score to identify candidate Mendelian disease genes, based on whole-exome sequencing of a small number of disease cases. We used whole-exome data for two Mendelian disorders for which the causal gene is known. Considering only genes that contained variants in all cases, the VEST gene score ranked dihydroorotate dehydrogenase (DHODH) number 2 of 2253 genes in four cases of Miller syndrome, and myosin-3 (MYH3) number 2 of 2313 genes in three cases of Freeman Sheldon syndrome.
Conclusions
Our results demonstrate the potential power gain of aggregating bioinformatics variant scores into gene-level scores and the general utility of bioinformatics in assisting the search for disease genes in large-scale exome sequencing studies. VEST is available as a stand-alone software package at http://wiki.chasmsoftware.org and is hosted by the CRAVAT web server at http://www.cravat.us
Related collections
Most cited references21

- Record: found
- Abstract: found
- Article: found
The UCSC Genome Browser database: update 2011
- Record: found
- Abstract: found
- Article: not found
Using mutual information for selecting features in supervised neural net learning.
- Record: found
- Abstract: found
- Article: not found