- Record: found
- Abstract: found
- Article: not found
Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations
Read this article at

Abstract
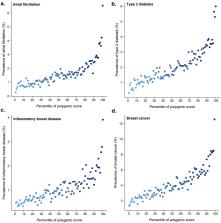
A key public health need is to identify individuals at high risk for a given disease to enable enhanced screening or preventive therapies. Because most common diseases have a genetic component, one important approach is to stratify individuals based on inherited DNA variation. 1 Proposed clinical applications have largely focused on finding carriers of rare monogenic mutations at several-fold increased risk. Although most disease risk is polygenic in nature, 2– 5 it has not yet been possible to use polygenic predictors to identify individuals at risk comparable to monogenic mutations. Here, we develop and validate genome-wide polygenic scores for five common diseases. The approach identifies 8.0%, 6.1%, 3.5%, 3.2% and 1.5% of the population at greater than three-fold increased risk for coronary artery disease (CAD), atrial fibrillation, type 2 diabetes, inflammatory bowel disease, and breast cancer, respectively. For CAD, this prevalence is 20-fold higher than the carrier frequency of rare monogenic mutations conferring comparable risk. 6 We propose that it is time to contemplate the inclusion of polygenic risk prediction in clinical care and discuss relevant issues.
Related collections
Most cited references11
- Record: found
- Abstract: not found
- Article: not found
Inflammatory bowel disease.
- Record: found
- Abstract: not found
- Article: not found
Rare and common variants: twenty arguments.
- Record: found
- Abstract: found
- Article: not found