- Record: found
- Abstract: found
- Article: found
Genomic Distribution and Inter-Sample Variation of Non-CpG Methylation across Human Cell Types
Read this article at
Abstract
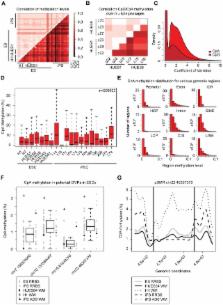
DNA methylation plays an important role in development and disease. The primary sites of DNA methylation in vertebrates are cytosines in the CpG dinucleotide context, which account for roughly three quarters of the total DNA methylation content in human and mouse cells. While the genomic distribution, inter-individual stability, and functional role of CpG methylation are reasonably well understood, little is known about DNA methylation targeting CpA, CpT, and CpC (non-CpG) dinucleotides. Here we report a comprehensive analysis of non-CpG methylation in 76 genome-scale DNA methylation maps across pluripotent and differentiated human cell types. We confirm non-CpG methylation to be predominantly present in pluripotent cell types and observe a decrease upon differentiation and near complete absence in various somatic cell types. Although no function has been assigned to it in pluripotency, our data highlight that non-CpG methylation patterns reappear upon iPS cell reprogramming. Intriguingly, the patterns are highly variable and show little conservation between different pluripotent cell lines. We find a strong correlation of non-CpG methylation and DNMT3 expression levels while showing statistical independence of non-CpG methylation from pluripotency associated gene expression. In line with these findings, we show that knockdown of DNMTA and DNMT3B in hESCs results in a global reduction of non-CpG methylation. Finally, non-CpG methylation appears to be spatially correlated with CpG methylation. In summary these results contribute further to our understanding of cytosine methylation patterns in human cells using a large representative sample set.
Author Summary
Epigenetic modifications including DNA methylation at the position 5 of the cytosine base provide regulatory information to the genome sequence. The primary target of cytosine methylation in mammals is the CpG dinucleotide. However, previous studies in the mouse and more recent work in humans have highlighted the presence of non-CpG methylation in pluripotent cells. Currently, little is known about the role of this type of DNA methylation. We sought to further characterize non-CpG methylation by employing a comprehensive data set of genome-scale methylation maps across various human cell types. Our analysis reveals that non-CpG methylation varies dramatically between pluripotent cells and is closely linked to CpG methylation. Moreover, we show that depletion of the de novo DNA methyltransferases results in a global reduction of non-CpG methylation levels. Taken together, these findings further advance our understanding of cytosine methylation and describe its distribution among a large number of human cell types.
Related collections
Most cited references23
- Record: found
- Abstract: found
- Article: not found
Ensembl 2009
- Record: found
- Abstract: found
- Article: not found
Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome.
- Record: found
- Abstract: found
- Article: not found